
PI3Kα
March 2024
PI3Kα
Phosphoinositide 3-kinase alpha (PI3Kα) is a lipid kinase frequently mutated in human cancer (14% of all solid tumors). PI3Kα mutants show enhanced activity compared to the wild-type (WT) protein, leading to dysregulation of a wide range of cellular processes that can drive cancer development.
PI3Kα consists of two subunits: a catalytic subunit known as p110α (PIK3CA) and a regulatory subunit, most commonly p85α. The cancer-causing mutations occur in the kinase p110α subunit, and they are not found at the catalytic, ATP-binding site (orthosteric site) but in other regions of the protein.
The development of PI3Kα inhibitors has thus far focused on targeting the kinase active site (orthosteric inhibitors), but these compounds show poor selectivity in inhibiting the mutated PI3Kα over the WT functional protein. The unselective inhibition of WT PI3Kα causes significant side effects, mainly associated with the alteration of glucose and insulin metabolism (hyperglycemia), that have limited the successful application of orthosteric PI3Kα inhibitors in the clinic.
This work mostly focused on one of the most frequently observed cancer-associated mutants, H1047R PI3Kα. Previous studies elucidated the 3D structures of this mutant and showed that the H1047R mutation disrupts the conformation of the p110α subunit C-terminal tail (where the mutated residue is located, Image 1). Importantly, this mutation does not significantly alter the structure of the kinase active site, indicating that the enhanced activity of the mutated protein is not associated with perturbations in the catalytic site but rather due to a conformational regulation.
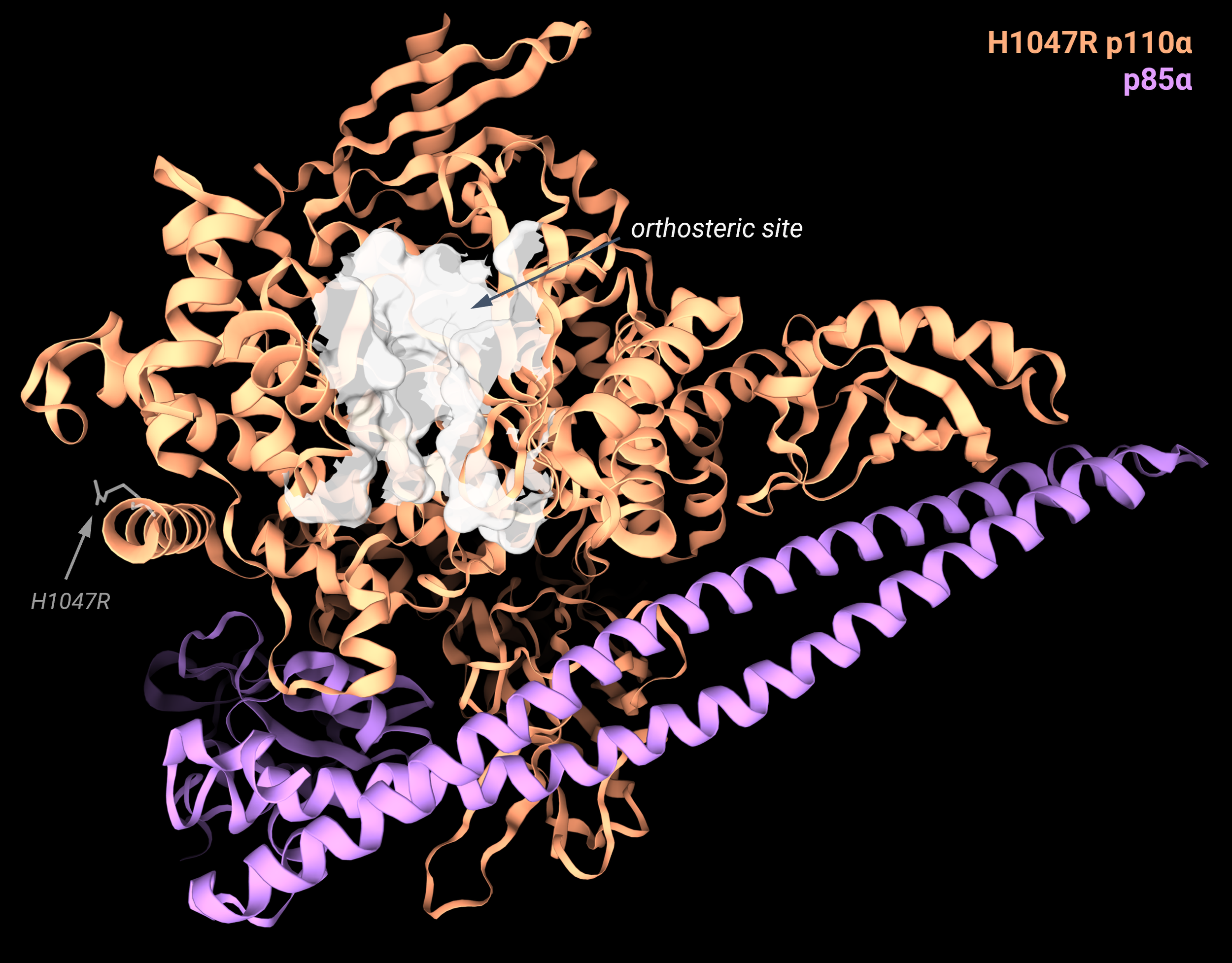
Image 1: Cryo-EM structure of the H1047R mutant (PDB: 8GUB), with the H1047R residue highlighted and labeled in grey. The p110α subunit is in orange, the p85α subunit in purple. The picture is produced with the 3decision® software.
Relay Therapeutics scientists performed molecular dynamics (MD) simulations to elucidate conformational dynamics differences between WT and H1047R PI3Kα. They confirmed the previous structural observations of high conformational mobility of the C-terminal tail for the mutant, but they also detected a slightly higher disorder of the activation loop in the mutant kinase domain than in the WT. With other MD simulations, they identified some crucial residues located in-between the C-terminal tail and the activation loop (F954 and D915). This showed altered conformations compared to the WT ones, indicating that they could play a crucial role in linking the site of mutation to the activation loop, constituting an allosteric network.
The scientists solved X-ray structures of WT and H1047R to validate their theory. In the WT protein, the tail had a buried conformation with the residues H1047 and W1051 interacting and the loop containing F954 protruding away from the C-terminal tail (Image 2A). The C-terminal tail was solved until the residue W1051. In contrast, the PI3Kα mutant C-terminal tail could only be solved until the mutated arginine (H1047R). The lack of electron density in this region of the mutant indicates higher flexibility because the arginine cannot engage in the same stabilizing interaction network as the WT histidine residue. In the H1047R PI3Kα, the F954 loop rotates towards the C-terminal tail to fill the space occupied by the H1047 in the WT structure (Image 2B). Therefore, a marked difference between the WT and the H1047R mutant in this region can be observed (Image 2C). From these structural considerations, the scientists hypothesized that targeting this allosteric network could inhibit the activity of the H1047R mutant while also offering mutant selectivity.

Image 2: Comparison of X-ray structures of WT p110α (PDB: 8TS7, orange) and H1047R p110α mutant (PDB: 8TS8, blue). A. Highlighted in white are the WT p110α conformations of residues: W1051, H1047, F594, and D915. B. Highlighted in white are the H1047R p110α mutant conformations of residues: R1047, F594, and D915. C. Superposition of the WT and H1047R p110α subunits, represented with the 3decision® highlight mode: the residues with an RMSD higher than 1.5 are displayed as lines and cartoon, while for the residues with a lower RMSD, only the backbone line is represented. This visualization mode allows easy spotting of the conformational differences between the residues composing the allosteric network between mutant and WT. All pictures are produced with the 3decision® software.
The researchers performed a DNA-encoded library screen and, from the hits, developed compound 2, which showed a 4-fold higher inhibition of the H1047R mutant over the WT. From the structural solution of the protein in complex with the ligand, they could assess the binding site and ligand pose (Image 3A), confirming the binding to the region they identified implicated in an allosteric regulation of the protein. The ligand displaces many residues of the allosteric network, causing a loss of function of the kinase catalytic activity and stabilizing an inactive conformation. Further optimization of compound 2 resulted in RLY-2608, with improved potency and selectivity (12-fold higher inhibition of the H1047R mutant ) due to a better space filling of the binding site, as elucidated from the 3D structure (Image 3B).

Image 3: Structures of the H1047R p110α mutant in complex with the novel allosteric inhibitors. 2D chemical structures of compound 2 and RLY-2608 are reported on the right. A. H1047R p110α complex with compound 2 (PDB: 8TSA, protein in white, ligand in grey). The electron density map of the X-ray structure is reported and shows a clear density, indicating the binding site and ligand pose of compound 2. B. Comparison of the binding poses of compound 2 (PDB: 8TSA, grey) and RLY-2608 (PDB: 8TSD, green). The pocket residues and surface of H1047R p110α are reported. The pictures are produced with the 3decision® software.
Clinical studies using RLY-2608 showed significant tumor growth inhibition with minimal side effects. This work exploited structural information and conformational dynamics analysis to design a safer PI3Kα inhibitor, introducing novel treatment possibilities for patients afflicted with PIK3CA-mutant tumors.