
Pf20S
April 2024
Pf20S
Malaria is an infectious disease caused by Plasmodium parasites, which pose a significant global health threat. According to the World Health Organization (WHO), around 249 million people were affected by malaria worldwide in 2022, and it caused around 608,000 deaths.
Despite advancements in antimalarial treatments, the widespread use of antimalarial medications has led to the emergence of drug resistance in malaria parasites, creating an urgent need for new therapeutic approaches.
The proteasome of the malaria parasite Plasmodium falciparum (Pf20S) is a multiprotein enzymatic complex involved in protein degradation within the parasite. Proteasome inhibition disrupts various stages of the parasite's life cycle, making Pf20S an advantageous antimalarial drug target.
Recently, proteasome inhibitors have been developed that showed potent antimalarial activity and synergy with current therapies. Together with potency, resistance occurrence was evaluated in the early stage of drug discovery for such novel antimalarial treatments to minimize the emergence of resistance in the clinical setting.
Scientists obtained the cryo-EM structure of Pf20S with the inhibitor TDI-8304 (PDB: 8G6E) at 2.18 Å resolution, allowing unambiguous determination of ligand binding. Previous biochemical studies showed that TDI-8304 effectively inhibits the β5 subunits of Pf20S. The electron density map confirmed that two TDI-8304 molecules interact with the two β5 subunits. Surprisingly, the experimental structure also revealed four additional TDI-8304 molecules: two at the β2 subunits and two in a pocket formed by subunits β3, β4, and α3 (Image 1A).
TDI-8304 binds the β5 subunit with an extensive interaction network, which explains its high inhibition potency of the β5 subunit activity. On the other hand, TDI-8304 only moderately inhibits the β2 subunit activity. This difference was found to be due to the pyrrolidinone moiety on TDI-8304, which forms strong interactions with the β5 subunit (such as two hydrogen bonds with Ser154, Image 1B), while analogous interactions are not present in the β2 subunit (Image 1C). These structural details help explain why TDI-8304 is less effective at inhibiting β2 than β5. The third location where TDI-8304 binds does not have a significant inhibitory effect on Pf20S.
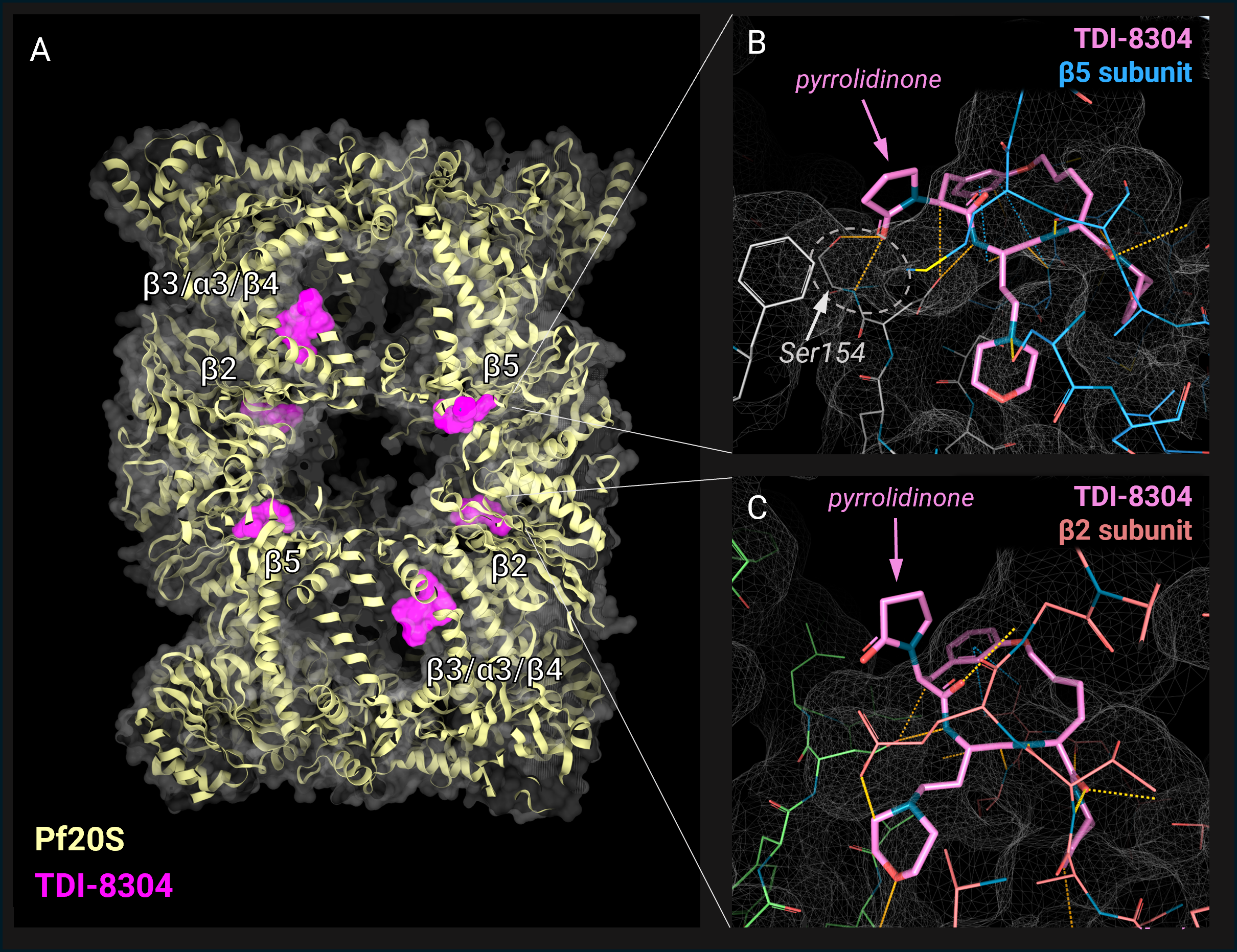
Image 1. TDI-8304 binding mode with Pf20S. A) Cryo-EM structure of Pf20S in complex with six TDI-8304 molecules (PDB: 8G6E). Pf20S cartoon representation in yellow and molecular surface in grey, ligand molecular surface in magenta. The subunits composing the protein complex are indicated. Surfaces are produced with the 3decision® software. B) Zoom on the TDI-8304 binding site in the β5 subunit. TDI-8304 in pink, β5 subunit residues in blue and the pocket surface is represented in white mesh. Pyrrolidone moiety on TDI-8304 and the residue Ser154 are indicated; hydrogen bonds between pyrrolidone and Ser154 are in the dotted circle. C) Zoom on the TDI-8304 binding site in the β2 subunit. TDI-8304 in pink, β2 subunit residues in mutton red, and the pocket surface is represented in white mesh. Pyrrolidone moiety on TDI-8304 is indicated.
Previous studies conducted on preclinical models have shown that P. falciparum acquires resistance to TDI-8304 by a single mutation at the Pf20S β6 subunit (A117D). However, this same mutation makes the mutant strain more sensitive to β2 inhibitors (collateral sensitivity). To find the structural rationale for this behavior, the researchers solved the 3D structure of the Pf20S β6A117D mutant with a β2 inhibitor, WLW-vs (PDB: 8G6F).
Interestingly, WLW-vs binds both the β2 and β5 subunits in Pf20Sβ6A117D. Comparing the structures of wild-type Pf20S and Pf20Sβ6A117D mutant, it was clear that the β6A117D mutation induced a significant conformational change in a region adjacent to the β5 binding pocket (Image 2). In the wild-type, the Tyr158 points away from the β5 subunit binding site and interacts with the Ala117. In the mutant β6A117D, the aromatic side chain gets repulsed by the negative charge of the Asp117, causing a flip of the Tyr158 residue and an overall flip of the entire Gly156-Ala161 region.

Image 2. Comparison of the structures of wild-type Pf20S with TDI-8403 (PDB: 8G6E; Pf20S in light grey, TDI-8304 in pink), and Pf20S β6A117D mutant (PDB: 8G6F; in blue). In the dotted grey circle are highlighted the residues at the 117 position: Ala117 in the wild-type (label in light grey), and Asp117 in the mutant (label in blue). The picture is produced using the 3decision® highlight mode: the residues with an RMSD higher than 2 are displayed as lines and cartoons, while for the residues with a lower RMSD, only the backbone line is represented. This visualization mode allows easy spotting of the conformational change induced by the point mutation β6A117D. The region Gly156-Ala161 is flipped in the mutant compared to the wild-type. All pictures are produced with the 3decision® software.
The conformation stabilized by β6A117D mutation limits the access of TDI-8304 to the β5 subunit binding site: the Tyr158 sterically clashes with the morpholine moiety of TDI-8304, and the Ser154 backbone moves away and loses the interaction with the pyrrolidinone moiety (Image 3A). This leads to a weaker binding of TDI-8304 to the β5 subunit, providing a structural explanation for the reduced β5 inhibition in the β6A117D mutant and the occurrence of resistance to the β5 inhibitor TDI-8304.
In addition, the new position of Tyr158 induced by the β6A117D mutation makes this residue available for interaction with the β2 inhibitor WLW-vs present in the β5 pocket through π- π stacking with the indole ring (Image 3B). This explains why this β2-specific ligand also binds to the β5 subunit in Pf20Sβ6A117D and supports the observed enhanced activity of this compound for the mutant.
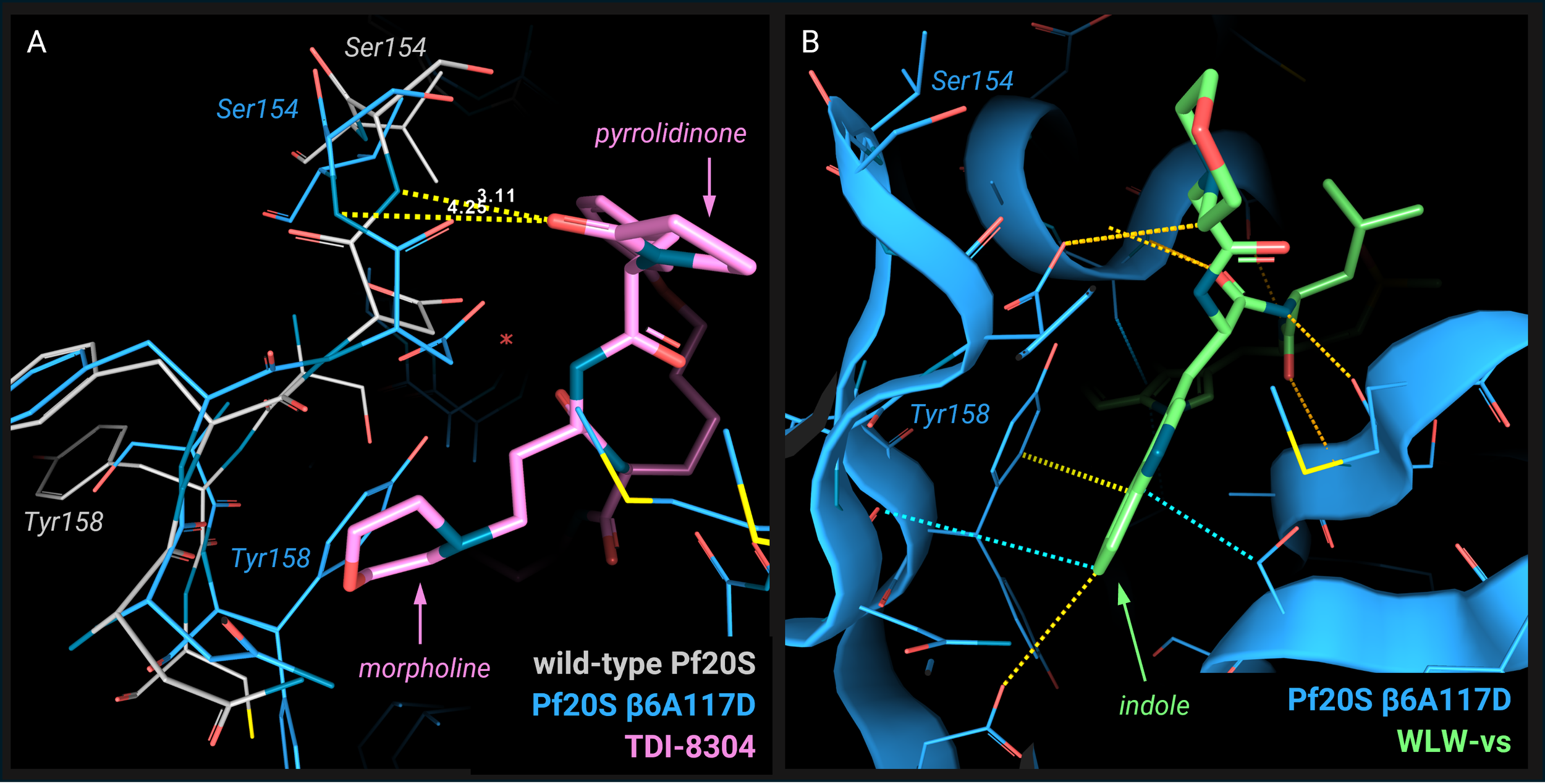
Image 3. A) Superposition of the TDI-8304 binding pose in the wild-type Pf20S (PDB: 8G6E; Pf20S in light grey, TDI-8304 in pink) with the Pf20S β6A117D mutant (PDB: 8G6F; Pf20S in blue). Residues Tyr158 and Ser154 are labeled in light grey for wild-type and in blue for mutant. The morpholine and pyrrolidinone moieties are indicated. The distance between the pyrrolidinone and the Ser154 NH backbone position in wild-type is 3.11 Å allowing a hydrogen bond, while the Ser154 in the mutant is 4.25 Å. The picture and measurements are produced using the 3decision® software. B) Pf20S β6A117D mutant β5 subunit pocket in complex with WLW-vs (PDB: 8G6E; Pf20S in blue, WLW-vs in green). Residues Tyr158 and Ser154 are labeled; the indole moiety is indicated. The pictures are produced with the 3decision® software.
The structural insights into the binding mode of proteasome inhibitors to Pf20S provide new opportunities for designing compounds that target both β2 and β5 subunits, which could lead to medications with lower risk of resistance emergence.
Reference:
Hsu, HC., Li, D., Zhan, W. et al. Structures revealing mechanisms of resistance and collateral sensitivity of Plasmodium falciparum to proteasome inhibitors. Nat Commun 14, 8302 (2023). https://doi.org/10.1038/s41467-023-44077-2