
5 fast-growing techniques in structural biology and their impact on drug discovery
Did you know that the first drug derived from a structure-based approach is used to treat high blood pressure ? That’s right, an antihypertensive agent called captopril was approved at the beginning of the 80s after researchers based their design on a theoretical structure of the target ACE.
Since then, science and technology have come a long way - many new processes have been incorporated to enable rational drug design driven by structural knowledge.
One field that has seen a remarkable advancement over the last decades is structural biology. Thanks to the great progress in 3D structure determination techniques, scientists can benefit from structural information of their biological targets now more than ever. Moreover, the field’s evolution is perfectly reflected in the open-access, 3D structural data repository Protein Data Bank (PDB). In the year in which the scientific community celebrates its 50th anniversary, PDB counts over 180,000 entries coming from many new technologies.
Fun fact: when the PDB was first announced in 1971 in the Nature New Biology journal, the community waved a red flag, saying that “The success of the proposed system will depend on the response of protein crystallographers supplying data.” Half a century later and more than the 25,000-fold increase in biomolecular entries, it turned out to be quite a success, didn’t it?
In this article, I will highlight 5 evolving structural biology techniques that are driving this ever-growing field and their impact on drug design and discovery, plus some interesting stats and a poll at the end.
1. Cryo-Electron Microscopy (Cryo-EM)
The “resolution revolution” of Cryo-EM brought this technique to the drug discovery scene.
Richard Henderson, 2017 Nobel Laureate, estimates that more protein structures will be determined by Cryo-EM than by X-ray crystallography in the next 5 years. And “as long as the number of the Cryo-EM structures increases, the interest and expectations from pharma are growing,” as mentioned in Jean Paul Renauld’s review, where he summarizes the latest progress in this technology.
Indeed, scientists have now a much broader access to structures of biomolecules that couldn’t be determined before, as Cryo-EM delivers new structural insights in challenging targets and provides an in-depth understanding of the mechanisms of diseases.
Currently, it has the greatest impact in the early stage of drug discovery, namely in the hit identification and target selection phases. It helps research teams to identify novel binding sites - orthosteric and allosteric - and to determine binding of the new small-molecule ligands. Some great examples of the successful application of Cryo-EM in pharma are listed here
*** Due to the high interest of our readers in the application of Cryo-EM in drug discovery, we organized an event with domain experts from pharma to elaborate on the topic ***
Interestingly, applying the electron microscopy (EM) method for imaging biological samples was considered nearly impossible until the introduction of sample flash-freezing and preservation in vitrified ice. The vitrification process was a real “plot twist” in the history of cryo-EM since this procedure enabled imaging of complex biomolecules in a close-to-native environment and thus expanded the application of cryo-EM in drug discovery. Another breakthrough happened when the detector technology and image processing advanced up to the point where cryo-EM structures could be revealed in near-atomic resolution.
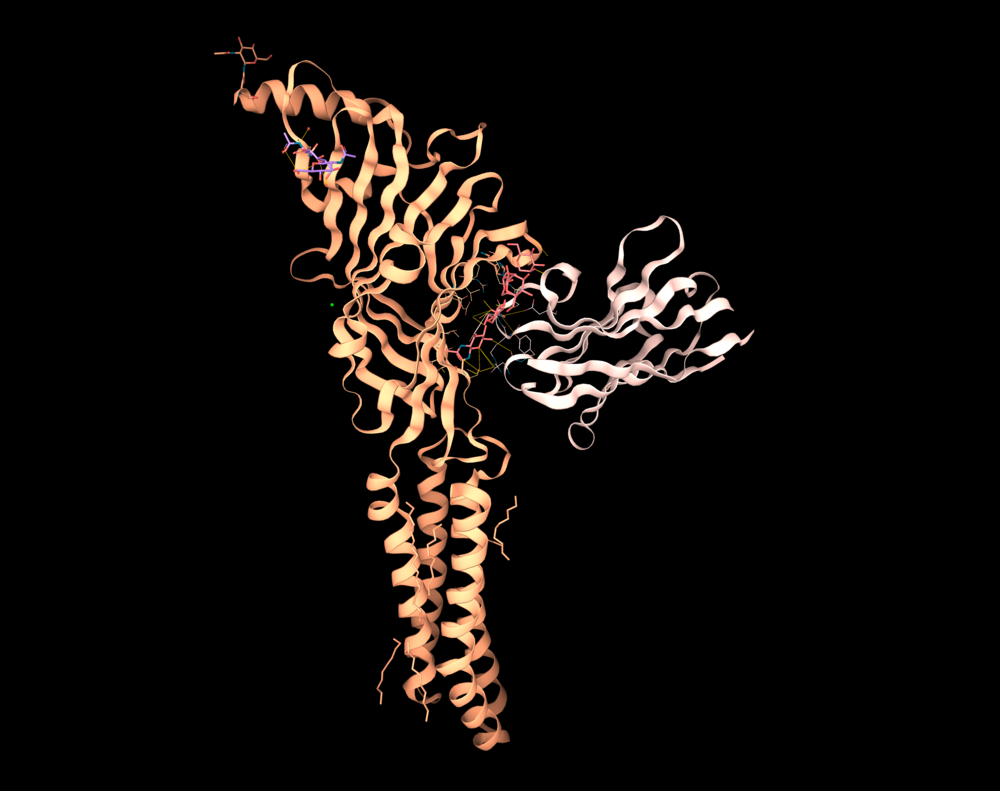
A GABAA receptor monomer: Cryo-EM resolved structure at atomic resolution, PDB ID: 7A5V.Image is captured in the 3decision 3D viewer.
In fact, the average resolution of a cryo-EM structure improved from 15 to around 6 Ångströms (Å) in the last 10 years. Nowadays many cryo-EM structures have a resolution of 3–4 Å. The most remarkable results count for those with sub-2 Å, like in the case of a GABAA receptor resolved at 1.7 Å (PDB ID: 7a5v).
Although Cryo-EM (here I refer to single particle analysis (SPA) technique) still comes with its own challenges and hurdles, it has several advantages when compared to the more traditional techniques (X-ray crystallography and NMR).
Cryo-EM does not require large amounts of sample nor the crystallization of the protein – an often time-consuming, and sometimes impossible challenge to tackle. These advantages enable scientists to capture large and more complex biological systems, membrane-bound systems, molecular chaperones, virus particles, bacteria, cells, and more. In addition, due to the significant improvements in image post-processing, cryo-EM makes it possible to determine multiple conformations of dynamic protein complexes.
One cannot discuss cryo-EM and its impact on drug discovery without mentioning microcrystal electron diffraction (MicroED) as well. This novel cryo-EM approach is now gaining momentum due to its ability to quickly characterize the structure of small organic compounds using nanocrystals, typically not suitable for X-Ray diffraction.
A MicroED structure determination workflow now allows to go from a compound sample powder to a high-resolution crystal structure within minutes.
Although improvements are still needed for cryo-EM to reach its full potential, like determining the structure of very small proteins, faster sample preparation, and lower price, the technique is on a very good way to further assist and accelerate the structure-guided drug discovery with its impressive progress.
Advantages of cryo-EM in a nutshell
-
Determination of large and/or dynamic molecules, such as membrane proteins
-
The formation of crystals is not required
-
Smaller amount of sample compared to X-Ray crystallography
2. High-Throughput (HT) Crystallography
Even though cryo-EM is the current “rising star” in structural biology, X-Ray crystallography still remains the number one technique for 3D macromolecular structure determination.
It is responsible for most of the structures stored in the PDB (almost 90%), and this number is growing each year, including for more complex systems (e.g. ribosomes). X-Ray crystallography is a very powerful tool for drug discovery as it can provide high resolution (now down to ≤0.7 Å) structures of protein-ligand complexes. It provides scientists with comprehensive structural insights for identifying druggable binding sites, or analyzing ligand binding modes, and generating rational drug design ideas.
During the last decades, the situation for X-Ray crystallography professionals changed a lot. The key factor was the drastic enhancement of the throughput thanks to extensive automation, brighter synchrotron X-Ray sources, increased power and speed of detectors, pipeline refinement, and other technical implementations.
These advances allowed experts to expand its application beyond mere structure determination towards downstream steps in the drug discovery pipeline, such as hit identification and lead discovery (even though it was not always easy nor practical due to the high amount of manual work, cost, and time required).
Contribution of X-Ray crystallography to early stages of drug discovery shown with crystal icons. Source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7179213/
It is now possible to determine macromolecular structures in less than a week and achieve the throughput of several protein-ligand complexes in just one day. Moreover, with high-throughput procedure scientists managed to extend the use of X-Ray crystallography even to primary screening purposes.
In fact, crystallographic fragment screening emerged as a very popular technique in the initial stages of fragment-based drug discovery (FBDD). With high-throughput X-Ray crystallography, one can analyze a whole ligand library from hundreds to a few thousand compounds in 1-2 weeks. With the aid of computer software (like PanDDA), the fragments with weak binding affinity and overlapping binding modes can be further refined. High-quality structures obtained this way can certainly facilitate the identification of fragment hits. Moreover, accurate structural information from fragment-protein complexes guides the follow-up studies on hit fragment evolution into larger molecules that are used as a starting point for lead discovery.
Despite these advancements, some limitations in X-ray crystallography that still persist are sample radiation damage and minimum crystal size and quality requirements to produce good results. The current efforts are further concentrated on improving validation, mining, and management of the big data coming from this technique for its comprehensive application.
Advantages of HT Crystallography
-
High-resolution of structures (even ≤0.7 Å)
-
Automation of data collection and refinement
-
Suitable for ligand screening campaigns in drug discovery (e.g., around 500 fragment-ligand complex data can be collected, processed and refined within 1 week; whereas the same process would take one and a half years with cryo-EM)
3. Serial femtosecond crystallography (SFX)
The hurdles in X-Ray crystallography mentioned above greatly limit the structure determination of some clinically important biomolecules
Membrane-bound proteins, for example, do not usually form enough large and pure crystals for the conventional crystallographic process. Despite their critical role in biological processes, only about 2% of the PDB entries are 3D structures of membrane proteins. The lack of structural information on these systems and other difficult-to-crystallize targets can hinder the development of efficient drugs.
Fortunately, certain progresses have been made to address these challenges: besides cryo-EM, the development of the X-Ray free-electron laser (XFEL) source is one of them. XFEL produces intense and ultrashort X-ray pulses (femtoseconds in duration) that generate a diffraction pattern before the radiation destroys the crystal. This concept is referred to as “diffraction before destruction.”
Consequently, serial femtosecond crystallography (SFX) has been established as a method that uses XFEL to collect series of datasets of many different crystals at room temperature.
This technique is therefore capable of revealing high-resolution structures of macromolecules that form tiny crystals, micrometers and nanometers in size, and/or those sensitive to radiation. Within a very short timeframe, SFX has become a powerful tool for determining hard-to-crystalize structures, including the aforementioned membrane proteins.
The first-ever crystal structure of the GPCR rhodopsin bound to arrestin was determined using SFX (PDB ID: 4ZWJ). This achievement facilitated the understanding of signal coupling for this complex to be used in rational small molecule discovery.
In addition to delivering novel structural knowledge, SFX facilitates time-resolved (TR) structural analysis. Scientists use it to study molecular processes that happen in one quadrillionth of a second - much faster than obtained with any other X-ray source.
Due to the very short beam and room temperature conditions, it is possible to capture protein dynamics and enzyme catalysis in real-time, and thus provide detailed insights into conformational states, mechanisms of reactions and intermediate reaction states that could not be observed before. Some of the first SFX-TR studies were described by Japanese scientists here.

SFX produced a series of snapshots of what conformational states the photoactive yellow protein (PYP) goes through after light absorption of the chromophore. Source https://pdb101.rcsb.org/motm/207
Although technically demanding and complex at present, the SFX shows great potential to enhance structure-based drug design studies thanks to very accurate structural details and biological mechanisms resolved using this technique.
Advantages of SFX
-
Lower sample damage - Ultrashort (femtosecond) X-ray pulses enable “diffraction before destruction” of individual crystals
-
Structural data obtained at room temperature
-
Usage of small crystals (nanometer to micrometer in size)
-
High-resolution dynamic structures – even in unstable intermediate states
Which one out of these structural biology techniques has the biggest potential to accelerate drug discovery?
4. Integrative/Hybrid (I/H) modeling
In addition to a single approach, the integrative structural biology technique is gaining momentum. It is because of its advantage to provide large, highly complex structures and heterogeneous assemblies, as well as to characterize protein dynamics, that cannot be achieved with a single technique alone.
In fact, the concept behind integrative structure biology is to combine data coming from experimental and computational methods to generate “hybrid / integrative” structural models. It has been used increasingly in the past few years since it keeps pace with the fast development of other structure determination techniques.
A reflection of this ramp-up is the creation of PDB-Dev, a public repository specifically dedicated to storing hybrid models. The prototype archiving system currently holds 72 entries and is meant to merge with the PDB in due course.
See November Protein of the Month where we described the novel structural insights on the phospholipase gamma based on the integrative structural biology approach.
Besides methods that give the whole picture of a structure at the (near)atomic level (X-ray, NMR, and Cryo-EM), there are also other techniques that provide complementary structural insights on biological systems. The combination of these data can greatly contribute to guiding modeling efforts. For example:
Cross-linking mass spectrometry (XL-MS) emerged as a powerful tool for providing data on the proximity and relative orientation of subunits within single or large protein complexes. It has become a method of choice for getting insights into protein-protein interactions and a reliable tool for capturing protein dynamics.
Small-angle X-Ray scattering (SAXS) can be applied on macromolecular complexes in solution without worrying about size limitations. SAXS can help determine the overall shape and size of the system (e.g. how many subunits are involved) but also give insight into conformational changes due to ligand binding. Progress in both data collection and analysis of SAXS profiles, allows the method to be involved in more and more studies with joint techniques.
Then, computational methods like docking, molecular dynamics, homology models are used to complete the picture of the large assembles.
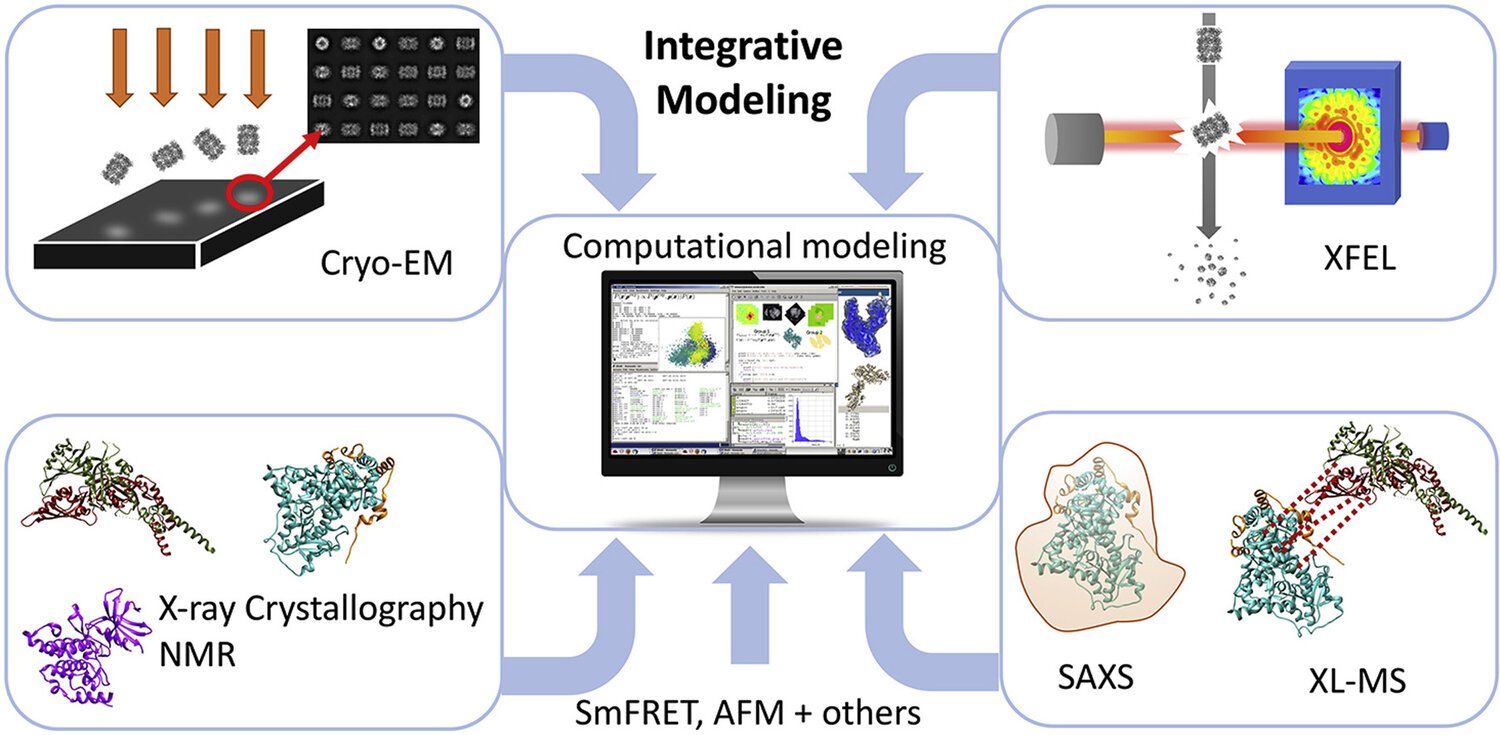
Illustration of hybrid modeling technique.
Source: https://www.sciencedirect.com/science/article/pii/S0022283620301054
Hybrid models are complex and require appropriate computer software to analyze results from different sources in a meaningful way. Besides technical requirements, advanced computational skills will be highly required as well for maximum potential.
This shift from one-technique-structures to those coming from a combination of methods and cross-discipline collaborations has a big potential to enable better understanding of entire cellular processes. Additionally, researchers will be able to rationally target large complexes, which nowadays is not simple.
H/I modeling main advantages
-
Structures of large and complex assemblies
-
Better understanding of biological pathways and disease mechanisms
5. In-silico protein structure predictions
Despite huge experimental efforts, only around 100.000 unique protein structures have been determined so far. It is, unfortunately, a trivial amount compared to the billions of known protein sequences. Therefore, computational methods are highly needed to reduce the protein-structure gap while complementing expensive and time-consuming laboratory setups.
Upon recently, the most successful protein structure prediction tools were based on homology modeling and fold recognition. However, with gaining experience in machine learning, an increase of computer power, and a lower cost of the hardware, a new complementary computational method has been developed.
In 2020, Google’s Deep Mind’s artificial-intelligence program AlpfaFold2 won the 14th Critical Assessment on protein Structure Prediction (CASP14) community-wide competition for the first time. The AlphaFold2 models outperformed others in the level of accuracy achieving a stunning median global distance test (GDT) score of 92.4 (out of 100).
Moreover, the program is able to deliver the structures very fast. DeepMind reported that computational models could be obtained within just an hour (depending on the structure size), which is faster than other existing methods.
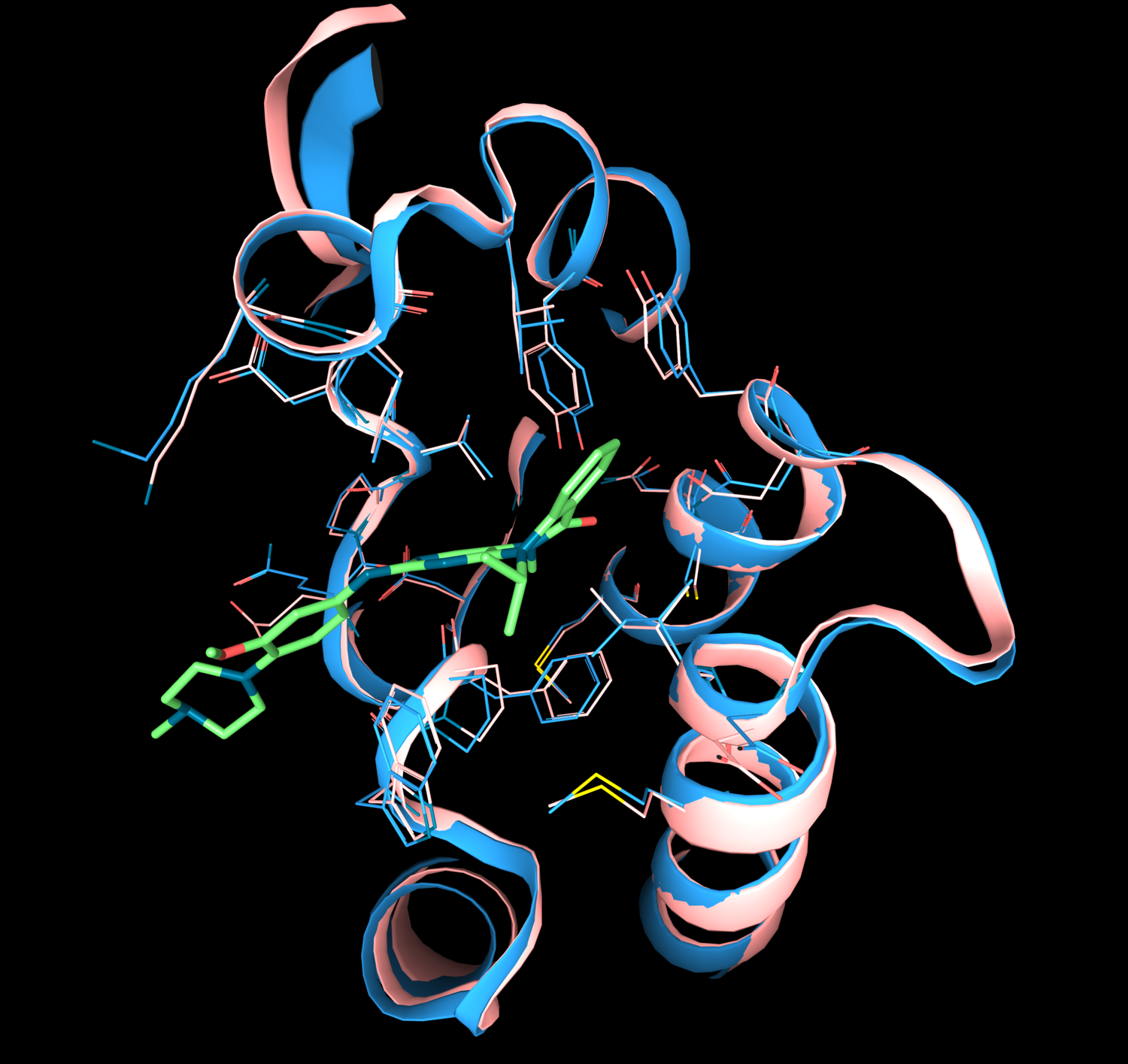
Comparison of experimental and theoretical structures of bromodomain-containing protein 4 (DeepMind AlphaFold model in pink and X-Ray structure (PDB ID:6CJ2) in blue) showing the accuracy of the AI model. The superposition was done with the 3decision software.
In July this year, DeepMind and EMBL-EBI established the AlphaFold protein structure database. All predicted structural models are now openly and publicly available for industry and academia. The DeepMind team plans to use their software to produce tens of millions of new structural models based on available sequences in the UniProt database.
While for scientists it is still early to predict the real-world impact of this breakthrough in life science, the potential for its application is huge, especially because it could improve efficacy and time management in drug discovery. AI prediction of still unavailable protein structures could be applied to better understand diseases, identify druggable sites, study protein-ligand interactions and ultimately develop new therapies (e.g. with protein design techniques).
In our previous article“Getting ready for AlphaFold with 3decision” we tackled some of the advantages and challenges of this new system from the data handling point of view.
So far, AI models have been especially useful for SarS-Cov-2 protein structures’ determination and have been applied in structure-guided vaccine design. This approach can then be extended to understanding future pathogens in a timely manner and get rapid therapeutic discovery.
Still, a couple of things are important to keep in mind when using AlphaFold2 models in a drug discovery context:
only structures of single protein chains have been predicted so far, no protein-protein complexes or complexes with oligonucleotides have been treated yet,
non-protein components (e.g., cofactors, ions, and ligands) are not included in the structure predictions. Initiatives such as AlphaFill have been started to enrich the models by modeling cofactors and ligands “back in” the protein.
Only one conformation of each protein domain has been modeled and published in the AlphafoldDB so the models do not reflect any of the system’s dynamics.
AlphaFold technology highlights
-
Predicted protein structures at a low cost (no experiments needed)
-
Fast structure prediction (completely new models can be produced in a few hours)
-
Known protein templates are no longer absolutely necessary
-
High accuracy of predicted models (comparable to experimental structures)
-
Freely and publicly available structure database (AlphaFold Protein Structure Database)
*** Update: The AlphaFold and ML-methods for protein structure prediction developed since the publication of this article. Have a look at our event Discngine Labs: “Protein Structure Prediction: What’s next after AlphaFold?" where experts from pharma and academia present the latest developments of deep-learning methods to support structural biology and drug discovery”. ***
Let’s talk numbers!
The impact of the fantastic progresses in structural biology on drug design and discovery is impressive.
In this paper, scientists summarize the success of FDA-drug approval related to structural knowledge in the period 2010-2016:
88% of approved drugs (184/210) had relevant protein structures stored in the PDB. The structures provided valuable information ranging from understanding biological mechanisms of the target to structure-guided drug discovery
95% of relevant structures were determined using X-Ray crystallography methods and the rest using NMR and electron microscopy
> 70% of anti-cancer small molecule drugs (45/59) approved during this period were products of structure-based drug discovery
These significant results highlight the wealth of information coming from each structure. And each new scientific and technical development in the field drive strategies for new discoveries.
It is not too late to give your vote!
We were curious to get the opinion of our community on this topic so we launched a LinkedIn poll asking them to choose one out of 4 proposed technics (Cryo-EM, HT Crystallography, SFX, and integrative modeling) that in their opinion will accelerate drug discovery the most in the near future.
Before scrolling down, try to guess which technique took the lead and get the chance to share the opinion on the same question by voting in this form. Plus feel free to choose the topic you like us to cover next.
