
HDAC
November 2022
Histone deacetylase (HDAC)
Histone deacetylases (HDACs) are a family of enzymes involved in the regulation of many genes, and so they are an attractive pharmacological target for a wide variety of diseases. Already five HDAC inhibitors have been approved for the treatment of some cancer types (T-cell lymphoma and multiple myeloma), and recently some more have also been evaluated for non-oncologic therapies (e.g. neurological diseases and HIV therapy).
Most of HDACs are structurally very similar and have a zinc-mediated catalytic activity. In order to suppress HDAC activity, inhibitors usually bind to the metal. However, the zinc-binding moiety of HDAC inhibitors causes several side effects, and scientists are looking for new ways to overcome this hurdle.
Recently, a group from Merck has reported the structure-guided discovery of a novel series of HDAC inhibitors, that preserve high potency without the zinc-binding motif.
They first studied the binding mode of the previously known inhibitor compound 7 with HDAC2. They found that 7 binds with the protein through three modalities (Image 1):
coordinates the zinc through a metal-binding moiety
forms an extensive network of hydrophobic and aromatic interactions thanks to its hydrophobic tail
interacts with the solvent-exposed surface of the protein with the remaining portion of the molecule
Image 1: On the left: molecular structure of compound 7, and description of the different chemical moieties (the molecule was drawn in the ChemAxon sketcher in 3decision®). On the right: 3D structure of the complex of 7 with HDAC2, interacting with the zinc ion (7 in white, HDAC2 in orange, zinc in light blue; PDB: 6WBW. Picture produced with 3decision® software).
From these structural insights, they decided to modify the surface-interacting portion of the molecule, by replacing the azetidine moiety with an indole, to increase the number of interactions with the protein. They reasoned that this modification could compensate for the removal of the zinc-binding moiety. In this way, they produced compound 19, which showed good potency. Then they further developed compound 22 (Image 2).

Image 2. Structure-guided optimization of HDAC inhibitors: in green is shown the modification for the removal of the zinc-binding moiety; in blue, the changes in a portion of the protein-surface interacting moiety; in orange, the final modification to put a polar group in the solvent-exposed part of the inhibitor.
When they compared the two structures of HDAC in complex with the compounds 7 and 19, they saw that the binding mode of 19 is not affected by the absence of the zinc-binding group and overlaps almost perfectly with 7 (Image 3A). As predicted, the indole in 19 forms a much more extended network of interactions with the surface of the protein, compared with the azetidine in 7 (Image 3B), thus compensating for the lack of interaction with the zinc.
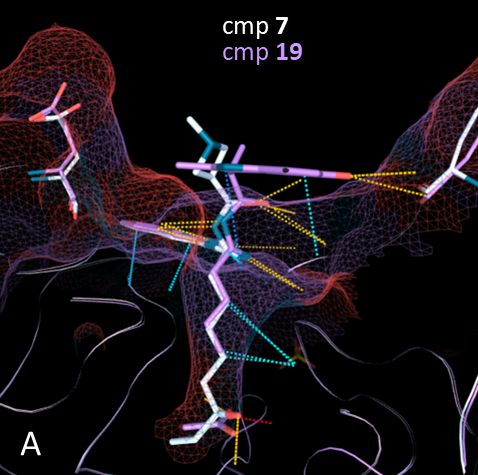

Image 3. A: comparison of binding modes of compounds 7 and 19 with HDAC2 (HDAC2:7 in white, PDB: 6WBW; HDAC2:19 in purple, PDB: 7ltl). The picture is made using the highlight mode of 3decision® software. B: detail of the protein surface binding mode for the two compounds. Note also the methyl protruding towards the solvent for compound 19.
Finally, they observed that the methyl on the methylene in 19 protrudes towards the solvent. In order to improve even more the potency, they added a polar amine group at this position, leading to compound 22 (Image 2), which showed properties comparable to marketed drugs.
In this way, the group managed to produce a proof-of-concept HDAC inhibitor, changing the classical paradigm that zinc-binding is pivotal to achieving high potency. All that is thanks to the crucial structural insights that guided the design and optimization of this novel series of compounds.
Reference:
Redefining the Histone Deacetylase Inhibitor Pharmacophore: High Potency with No Zinc Cofactor Interaction, Douglas C. Beshore, Gregory C. Adam, Richard J. O. Barnard, Christine Burlein, Steven N. Gallicchio, M. Katharine Holloway, Daniel Krosky, Wei Lemaire, Robert W. Myers, Sangita Patel, Michael A. Plotkin, David A. Powell, Vanessa Rada, Christopher D. Cox, Paul J. Coleman, Daniel J. Klein, and Scott E. Wolkenberg; ACS Medicinal Chemistry Letters 2021 12 (4), 540-547; DOI: 10.1021/acsmedchemlett.1c00074