
The impact of AlphaFold in drug discovery and emerging ML-methods
In the scope of drug discovery, 3D protein structures provide a unique source of information at many stages of drug development – from understanding the disease to studying protein-drug interactions, and ultimately developing new therapies. Even if more and more experimental protein structures (around 200.000) are being produced thanks to great improvements in protein structure determination techniques, only a limited number of proteins are represented in the PDB (55.000). For the protein families lacking experimental 3D structures, in silico predictions can be exploited to obtain structural information.
In 2020, AlphaFold (AF), a deep learning method developed by DeepMind, set a milestone in the field of protein structure prediction; It achieved impressive results in rapidly and accurately predicting 3D protein folds from the primary sequence alone, thus addressing the fundamental “protein folding problem”. The AF algorithm has so far predicted a huge number of protein structure models (over 200 million), providing scientists with unprecedented structural knowledge which has the potential to accelerate drug discovery.
However, despite this great accomplishment, the 3D protein structure alone does not tell the whole story. The functional context of AlphaFold models such as their arrangement with non-protein molecules, interactions with drugs, and dynamics, is still missing in the predictions. The scientific community is now using this technology as a starting point to develop methods that predict protein structure models more suitable for drug discovery applications.
In this blog post, we will give an overview of the real-life usage of AlphaFold models in drug discovery projects in the past two years. Also, we provide examples from pharma and academia of the latest developments of deep-learning methods to support drug discovery, from our Discngine Labs event on the topic: “Protein Structure Predictions: What’s next after AlphaFold?”.
AlphaFold usage in drug discovery: our poll
Even if the AlphaFold predictions are undoubtedly a breakthrough for fundamental research and protein science, their real contribution to drug discovery is still to demonstrate. After more than one year since their first release, we - at the 3decision team - wondered if and how AlphaFold has started to make a difference in drug discovery. We conducted a LinkedIn poll and a “live” poll during our latest Discngine Labs on this topic, asking a very simple “yes or no” question to our scientific community: “Have AlphaFold models already helped you significantly in a drug discovery project?”
This is the result we obtained from both polls: out of 240 scientists voting, around 1/3 of the participants have successfully used AlphaFold to support drug discovery campaigns, while the majority have still not experienced any useful impact.
Have AlphaFold models already helped you significantly in a drug discovery project?

Results of the poll “Have AlphaFold models already helped you significantly in a drug discovery project?” that was conducted on LinkedIn (61 votes) and live during the Discngine Labs event (178 votes). The poll participants are mainly scientists from pharma and biotech, working in the early stages of drug discovery. The results from both polls are plotted together since there were no significant differences in the percentages obtained from the two different sources (LinkedIn: yes 34%; DNG Labs: yes 38%).
On the “yes” side, we investigated further in which way the models positively contributed, and we found that AI-predicted structures have been mostly used for:
Validating a biological target (druggability assessment)
Supporting the solving of new protein structures (especially for cryo-EM)
Determining the function of an unknown protein
If AlphaFold is already making a difference when it comes to proteins for which we lack previous structural and functional information, the general feeling is that the predicted models are still far away from “competing with experimentally determined structures”, as sometimes we have - maybe too optimistically - heard in the past few months.
On the other hand, most scientists report that despite the dramatic development of deep learning-based technologies for structural prediction, there are strong limitations when it comes to their application to drug discovery problems. From the “no” side of our poll, we got feedback that AF still too often provides poor structure predictions, especially for “hard-to-solve” regions, and that sometimes the predicted models are not completely biologically relevant. As our event chair, Seth Harris from Genentech, commented:

"AlphaFold revolutionized how protein structure predictions are done. But AF only predicts one fold, so how do we get from there to understand the conformational dynamics or small-molecule binding?"
- Seth Harris, Director of Computational Structural Biology, Genentech
However, every day we read new reports of exciting improvements in AI-based methods that are addressing these limitations and are rapidly pushing these technologies closer to their application in “real-life” issues of drug discovery.
Insights from our Discngine Labs
In our fourth Discngine Labs, we have gathered scientists from academia, pharma, and biotechs, to present some of the latest advancements in machine-learning-based methods applied to drug discovery issues and their integration into the structure-based drug discovery workflow.
In the following sections, we summarized the main take-away messages from each presenter, who focused on one specific issue on the AF application to drug discovery. At the bottom of this page, you will find the link to the recording of the event.
Predicting co-factors and small molecule binding
Robbie Joosten, from the Netherlands Cancer Institute, treated one major problem of AF models: they are “protein only”, so they do not contain any of the compounds that are crucial for protein function, such as metal ions, ligands, and co-factors. To enhance the biological interpretation of the predicted structures, Robbie and his team developed AlphaFill, a homology-based algorithm that enriches AF models by “transplanting” co-factors that have been experimentally observed in homologous protein structures into the predicted one.
AlphaFill models have been successfully validated against experimental structures and can now be exploited by scientists to gain more functional insights from the AF models. For instance, they can be used to identify metal-binding sites and to assess the biological state represented in the AF model (for instance active/inactive state of kinases).
However, since the AlphaFill model relies on homology, it can only be applied to AF models for which an experimental structure of a homolog is available. Therefore, they are now implementing an AI-based algorithm to predict ligand-binding pockets, just from the structure model alone. They started with the prediction of metal-binding sites, from which they have obtained promising results by the validation against experimental structures, and they are now actively pursuing the prediction of binding sites of larger, more complex systems.
"The AlphaFill databank was designed to help life scientists to easily generate new hypotheses for protein function and formulate relevant research questions."
- Robbie Joosten, Research associate, Netherlands Research Institute

Protein conformational ensembles predictions
Christian Tyrchan presented his work at AstraZeneca addressing another current pain point of AF models: only one protein fold is predicted, even if proteins exist in multiple conformational states with different biological relevance and function. To overcome this limitation and obtain an understanding of protein dynamics, Christian and his team are exploring the ability of deep learning (DL) methods to predict protein conformational space.
Using DL-predicted models (e.g. AlphaFold and trRoseTTA) as a starting point, they applied a DL-based method for conformational sampling of protein states. Their predictions confirmed the experimental structural dynamics for some of the use cases they selected for validating their method, with some limitations for proteins with metal ions and co-factors influencing the conformational equilibrium. Also, they observed that using their DL method, they obtained results comparable with Molecular Dynamics (MD) simulations, but in a matter of hours instead of the days or weeks that MD simulations required.
So, although the DL algorithm had been trained on static PDB structures, it was able to reproduce the protein flexibility to a certain extent. This study opens the way for further development in this area, which could significantly support drug discovery by providing a much clearer understanding of protein dynamics from the beginning of the drug discovery effort.

"An accurate prediction of protein conformational ensembles would help to prioritize drug targets or modalities and identify cryptic pockets and allosteric interaction to drive rational design."
- Christian Tyrchan, Team leader of Computational Chemistry, AstraZeneca


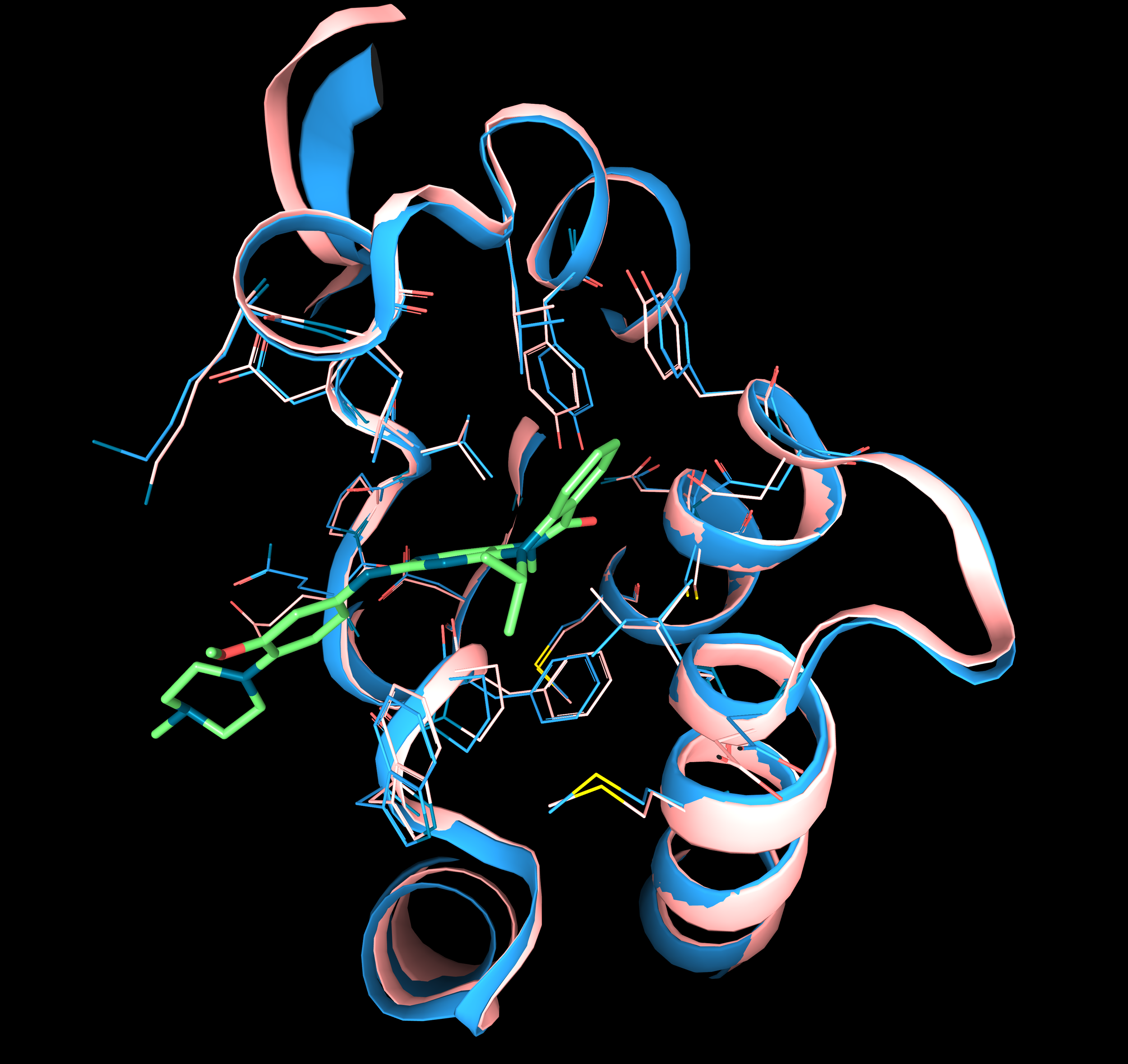
AlphaFold models and protein conformations. AlphaFold can only predict a single state of the protein conformation, so the model can either be representative of a single conformational state or be an average. For instance for Tyrosine-protein kinase Lck activation loop (on the left) if we compare the AF prediction (in yellow) with the experimental structures of different conformational states stabilized by two ligands (inactive, imatinib, in purple, PDB: 2PL0; active, imadizo purazin 1, light pink, PDB: 2ZM1), we observe that the model is not representative of either the states. On the contrary, for Adenylate kinase (on the right) AlphaFold very accurately predicts only one conformation: the AF model (in yellow) very precisely matches the experimentally determined structure of the close state of the protein (in light blue, PDB: 1AKE) and not the open (in dark blue, PDB: 4AKE). The superposition of structures is done with the 3decision® software.
Supporting protein crystallization
Jola Kopac showed how at Evotec, they are exploiting machine learning methods to support them in one of the major bottlenecks of any SBDD project: producing good quality protein crystals for obtaining structural information and driving drug design. It usually takes a lot of time and resources to obtain protein constructs that successfully crystallize, and sometimes, they do not. Jola and her team wondered if they could use ML methods to help them produce easy-to-crystallize protein constructs. They combined AlphaFold and another deep leaning-based method, ProteinMPNN (developed by David Baker’s Lab), which can be exploited for protein sequence design: given a protein backbone structure, it predicts the sequence that folds into this structure.
Starting from the expected fold of the target protein (from experimentally available structures or AF models), they exploited ProteinMPNN to optimize the sequence and provide them with an easy-to-crystallize protein construct. Then, they used AlphaFold to confirm that the optimized sequence still retained the original folding and did not alter critical regions for the biological activity, such as the binding site. For the biological targets they evaluated this method with, they never observed any difference in the binding site after the sequence optimization process. They are currently evaluating this pipeline in the wet lab and obtaining very promising results (still undisclosed).
"We developed a protocol that exploited ML methods to produce good crystal systems and rescue SBDD projects that previously failed due to lack of structural knowledge."
- Jola Kopac, Senior Scientist I, Evotec

Predicting protein-ligand complexes folding
Andrew Doré and Sergey Bartunov presented the core technology that drives drug discovery projects at CHARM Therapeutics: DragonFold, their (proprietary) deep learning-based algorithm which predicts protein/ligand co-folding. DragonFold predicts the full 3D structure for protein-ligand complexes using only the primary sequence of the target protein and the ligand’s chemical structure as inputs, with impressively high accuracy and in a matter of seconds. The algorithm predicts the ligand-induced protein fold, the ligand pose, but also the location of the binding site, which is automatically identified without additional inputs.
DragonFold provides fundamental help at the early, crucial stages of their drug discovery campaigns. Except for the obvious advantages of having structural information available from the very beginning of the project, DragonFold provides powerful support at the high-throughput-screening stage: since it can estimate the ligand affinity towards the protein, it can be exploited for hit finding, exploring a vast chemical space at very low cost, and with superior performances compared to virtual screenings. It can also detect small changes in the chemical structure of ligands and be successfully applied for lead optimization. Finally, it is being extensively used for selectivity studies and off-target predictions.

"DragonFold is our protein/ligand co-folding algorithm, which is driving the discovery of novel transformational medicines for hard-to-drug cancer targets."
- Andrew Doré, Head of structural sciences, Charm Therapeutics
Conclusion
The scientific community has witnessed major improvements in structural biology in the past few years, both in the experimental and in silico production of protein structures, that could drive the development of safer and more effective rationally designed drugs.
So far, AF models are not often used with success in drug discovery projects (as pointed out by our poll), but we can expect this to change very soon. From the recent literature - and all the great talks from our speakers - we can see that the key issues that limit the successful application of AF predictions in drug discovery are already being addressed by many ML methods currently under development. So deep learning methods (in general) are entering drug discovery workflows already today: in the presentations of our Discngine Labs, we could see that more and more ML-supported projects are being included in early drug discovery pipelines.
Even if this is just the beginning, and deep learning algorithms still need to prove their value in solving critical drug discovery problems, it is evident that they will be key players in the near future. We expect that in a couple of years if we asked again our poll question, the “yes” side would be much more numerous.
"10-15 years ago people did not even bother looking at computational models because they were wrong most of the time. But now, they are right!"
- Seth Harris, Director of Computational Structural Biology, Genentech
Want to know more?
If you enjoyed the article and the insights we collected on the application of AF in drug discovery and are interested in knowing more about the mentioned predictive methods, watch the recording of the Discngine Labs: “Protein Structure Predictions: What’s next after AlphaFold?” for free.
References:
Hekkelman, M. L.; de Vries, I.; Joosten, R. P.; Perrakis, A. AlphaFill: Enriching AlphaFold Models with Ligands and Cofactors. Nat. Methods 2022 202 2022, 20 (2), 205–213. https://doi.org/10.1038/s41592-022-01685-y.
Audagnotto, M.; Czechtizky, W.; De Maria, L.; Käck, H.; Papoian, G.; Tornberg, L.; Tyrchan, C.; Ulander, J. Machine Learning/Molecular Dynamic Protein Structure Prediction Approach to Investigate the Protein Conformational Ensemble. Sci. Reports 2022 121 2022, 12 (1), 1–17. https://doi.org/10.1038/s41598-022-13714-z.
Dauparas, J.; Anishchenko, I.; Bennett, N.; Bai, H.; Ragotte, R. J.; Milles, L. F.; Wicky, B. I. M.; Courbet, A.; de Haas, R. J.; Bethel, N.; Leung, P. J. Y.; Huddy, T. F.; Pellock, S.; Tischer, D.; Chan, F.; Koepnick, B.; Nguyen, H.; Kang, A.; Sankaran, B.; Bera, A. K.; King, N. P.; Baker, D. Robust Deep Learning–Based Protein Sequence Design Using ProteinMPNN. Science (80-. ). 2022, 378 (6615), 49–56. https://www.science.org/doi/10.1126/science.add2187
Technology - CHARM Therapeutics. https://charmtx.com/technology/ (accessed 2023-02-15).